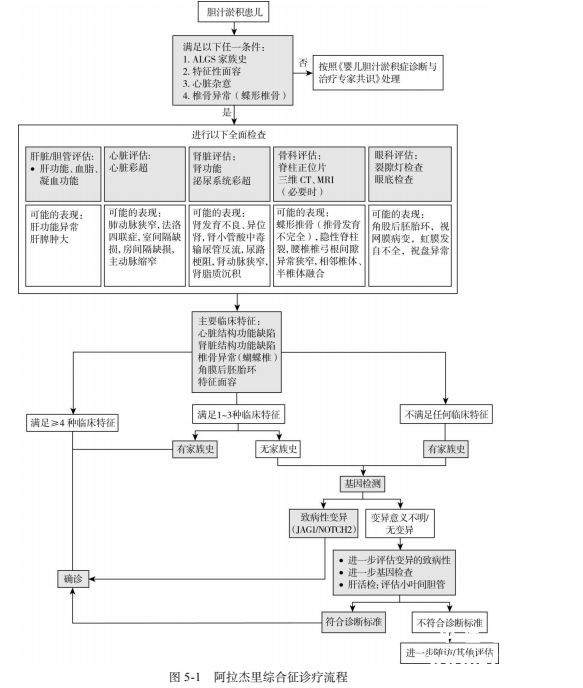
诊断 临床诊断的确立依赖于综合判断。经典的诊断标准为肝组织活检有肝内小叶间胆管数量减少或缺如,并具有至少包括慢性胆汁淤积、心脏杂音、蝴蝶椎骨、角膜后胚胎环和特殊面容等 5 个主要临床表现的其中 3 个,并排除其他可能原因。现在肾脏异常也列为主要异常之一。如果肝活检不表现为肝内小叶间胆管数量减少或缺如,或由于某些成年轻症病人并未进行肝活检,修订的诊断标准认为符合 4 个或以上主要标准也可诊断。如果已知有JAG1/NOTCH2 基因突变或阳性家族史时,2 个主要标准通常即可确诊。
鉴别诊断 ALGS患者血清GGT升高明显,因此需要和伴有GGT升高的各种婴儿期胆汁淤积症相鉴别。由于 ALGS 患者脊椎、眼睛和肾脏异常多无显著的临床表现,特征性面容在婴儿早期也不显著等原因,ALGS 与其他原因引起的胆汁淤积症鉴别也有一定难度。ALGS早期诊断面临的最大挑战是如何与胆道闭锁相鉴别。由于胆道闭锁需要尽早手术治疗,而有报道若把 ALGS 误诊而进行葛西手术可使预后变差,因此及时进行鉴别诊断尤为重要。婴儿高 GGT 胆汁淤积症患儿行脊柱摄片、心脏超声及眼科检查有助于鉴别 ALGS 与胆道闭锁。此外,肝穿刺组织活检对鉴别诊断也有很大帮助。胆道闭锁的特征是小胆管显著增生,而ALGS 虽然在早期可不存在肝内胆管消失或减少,但也少见显著小胆管增生。其他需要鉴别的以多系统受累疾病包括威廉姆斯综合征、歌舞伎(Kabuki)综合征等。
治疗 目前尚无特异的治疗手段,主要是支持治疗和对症处理。由于该病有多系统受累,因此良好的管理需要多学科专科医师参与。肝病方面主要面临的是胆汁淤积及其并发症,包括脂溶性维生素缺乏。通常需要常规补充脂溶性维生素,并通过维生素浓度或凝血酶原时间进行检测。利胆药可选用熊去氧胆酸,每天 20~30mg/kg,分次口服。最新批准的氯马昔巴特可用于治疗 1 岁及以上阿拉杰里患者的胆汁淤积性瘙痒,推荐剂量为 380µg/kg,每日一次,早餐前 30 分钟口服。应避免和胆汁酸结合树脂同时使用,或前后至少间隔 4 小时。建议使用前详细阅读说明书。 ALGS 病人需要定期监测肝功能变化,必要时肝移植。肝移植是 ALGS 终末期肝病患儿的有效治疗措施。
诊疗流程(图
5-1)25
参考文献 [1] VANDRIEL SM, LI LT, SHE H, etal. Natura l history
of liver disease in a large international cohort of children with Alagille
syndrome : re sults from the GALA study-Hepatology, 2023, 77(2):512-529. [2] LI L, DONG J, WANG X, etal. JAG1 mutation spectrum
and origin in Chinese children with clinical features of Alagille syndrome .
PLoS ONE, 2015, 10(6): e0130355. [3] GEISLER F, NAGL F, MAZUR PK, etal. Liver-specificina
ctivation of Notch2, but not Notch1, compromises intrahepatic bile duct development
in mice . Hepatology, 2008, 48(2):607-616. [4] CARPENTER CD, LINSCOTT LL, LEACH JL, etal. Spectrum
of cerebra l a rterial and venous abnorma lities in Alagille syndrome . Pediatr
Radiol,2018,48(4):602-608. [5] WANG JS, WANG XH, ZHU QR, etal. Clinical and pathological
cha racteristics of Alagille syndrome in Chine s e children. World J Pediatr,2008,4(4):283-288. |